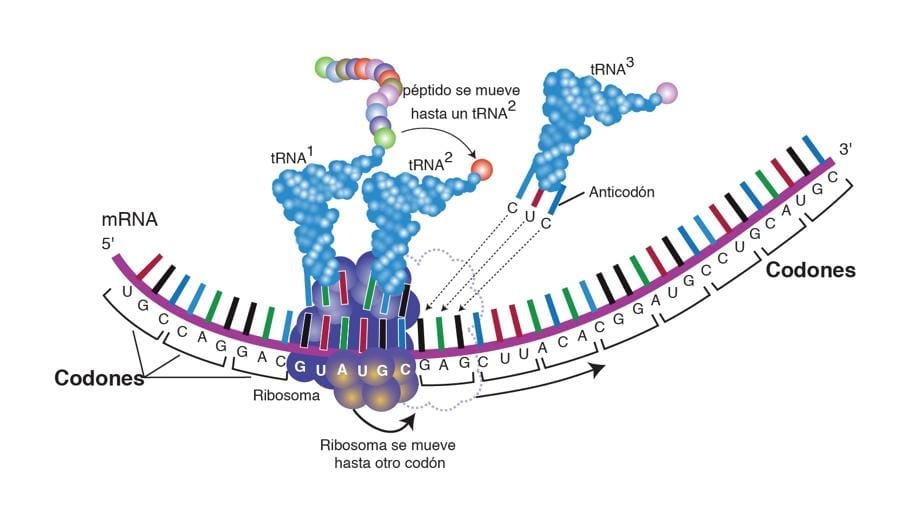
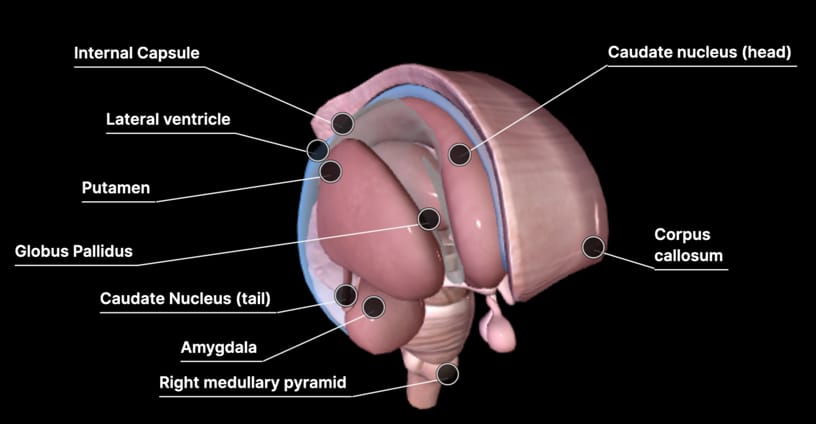
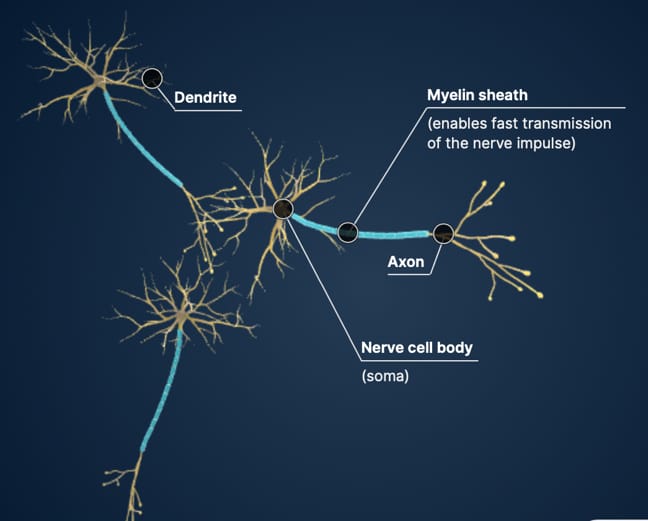
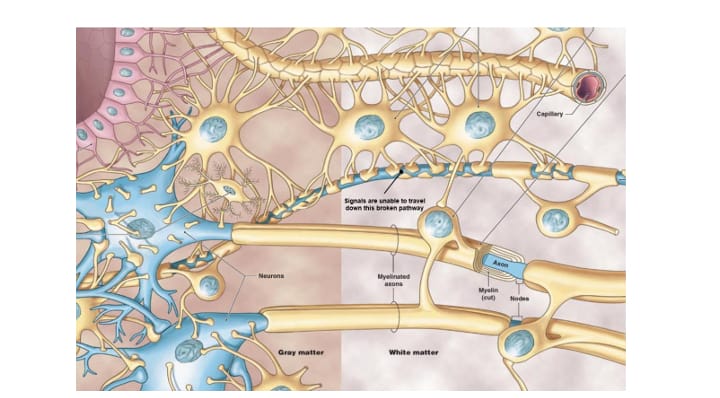
Las leucodistrofias se definen actualmente como todos los trastornos de base genética que afectan principalmente a la sustancia blanca del sistema nervioso central, independientemente del componente estructural involucrado, el proceso molecular afectado y el curso de la enfermedad.
Son enfermedades neurodegenerativas y los síntomas aparecen, en el 90% de los casos, en la infancia. La evolución de estas enfermedades acaba en muerte prematura del individuo afectado y la progresión depende de los genes involucrados.
La incidencia, en conjunto, se estima en 1 de cada 7000 individuos.
El aspecto más destacado es la ausencia o regresión en el desarrollo de un individuo que previamente parecía sano. Esta pérdida gradual puede manifestarse con problemas cognitivos, en el tono muscular, trastorno de movimiento, marcha, habla, capacidad para comer, visión, audición, cambios de comportamiento…
Actualmente hay identificados más de 50 tipos de mutaciones, apareciendo nuevas mutaciones (una o dos por año) por la efectividad del diagnóstico genético y el avance tecnológico.
Hay importantes avances en el campo de la investigación pero, actualmente, no hay cura posible para este espectro de síndromes, simplemente se tratan los síntomas con más o menos acierto.
Los individuos afectados, en su mayoría, son totalmente dependientes.
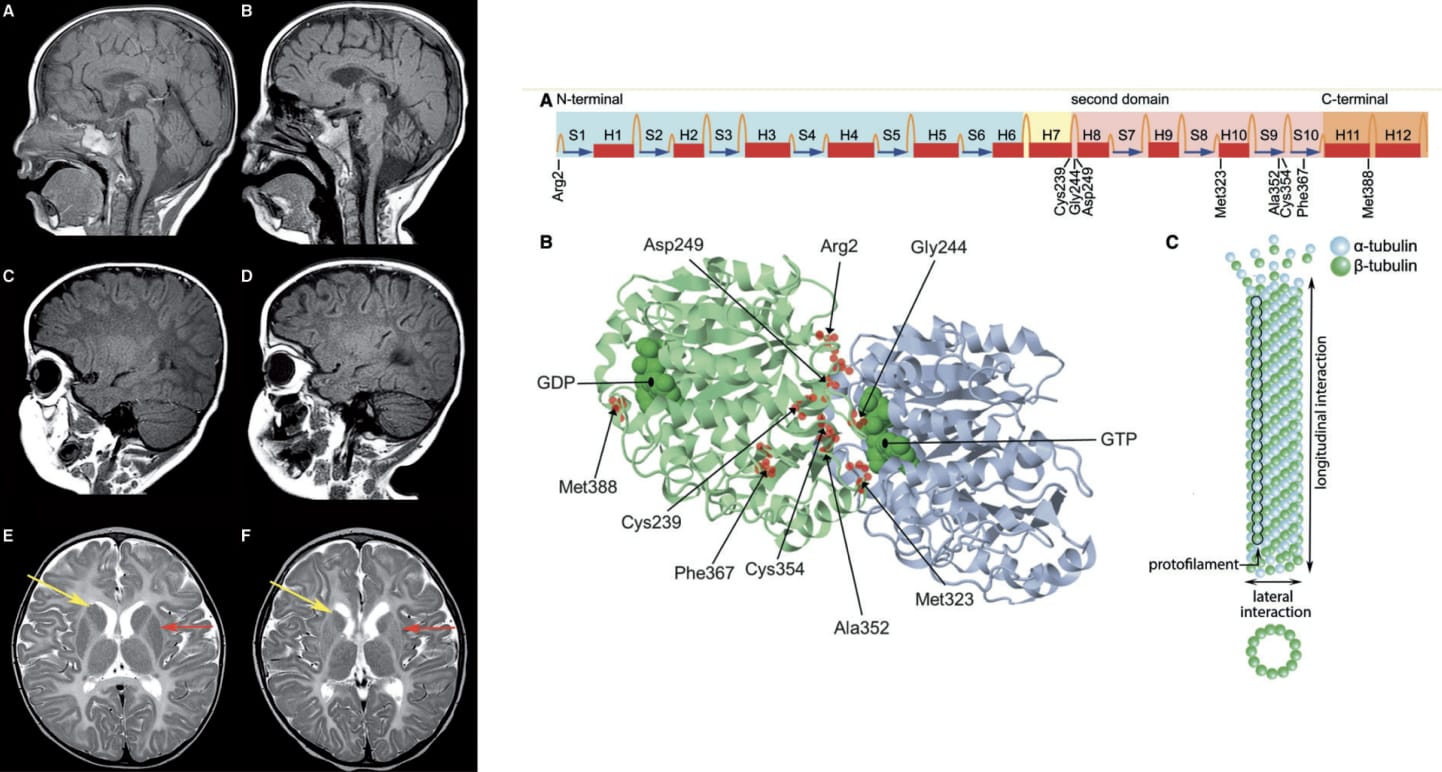
En la Leucodistrofia asociada al gen TUBB4A, las mutaciones tienen un carácter dominante.
La expresión del gen TUBB4A es total en el Sistema Nervioso Central (SNC) para la codificación de la proteína Beta -Tubulina, que forma los microtubulos. Estas estructuras son vitales para el desarrollo del SNC: las neuronas y las células gliales están afectadas.
Debido a las mutaciones, los microtúbulos no pueden hacer correctamente su función (incorrecta estabilización de la unión de las proteínas α- y β- tubulina) y la mielina no se puede disponer correctamente en los axones desde los oligodendrocitos, con lo que existen zonas no protegidas por esta “vaina” y, en consecuencia, los impulsos nerviosos están afectados desde su origen.
Además, genera un transporte intracelular ineficiente con el consiguiente efecto sobre la formación de neuritas y las conexiones neuronales.
La degeneración se produce por acumulación de proteínas no funcionales en neuronas y células gliales y, consecuentemente, muerte celular.